Macrophages Lacking TSC2 have mTORC1-dependent GPNMB Augmentation Ameliorating Cardiac Ischemia-Reperfusion Injury
Macrophages Lacking TSC2 have mTORC1-dependent GPNMB Augmentation Ameliorating Cardiac Ischemia-Reperfusion Injury
Keykhaei, M.; Koleini, N.; Meddeb, M.; Tajdini, M.; Rezaee, M.; Huang, Q.; Panesar, T.; Adamo, L.; Ranek, M.; Kass, D. A.
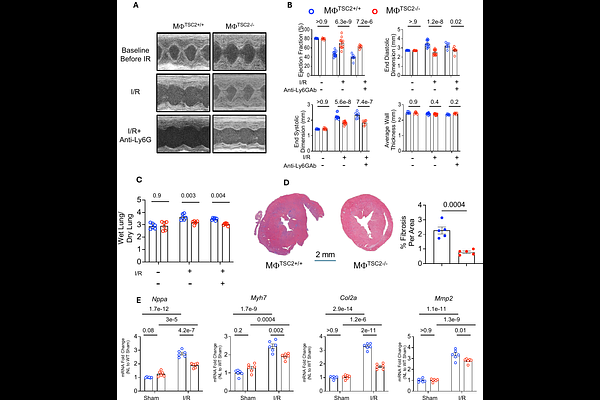
AbstractIntroduction: Macrophages (M{Phi}) modulate both myocardial inflammatory and reparative phases following ischemia-reperfusion (I/R) injury. The mechanistic target of rapamycin (mTOR) is thought to play an important role in M{Phi} phenotype and functionality, but studies report conflicting net influences suggesting dependence on disease context and downstream signaling. Here, we tested the impact of M{Phi} with constitutive mTORC1 activation induced by targeted deletion of tuberous sclerosis complex 2 (TSC2) on cardiac responses to I/R injury. Methods/Results: Myeloid TSC2 depleted (M{Phi}TSC2-/-) mice were generated by crossing Lys2Cre x TSC2flx/flx. Bone-marrow derived M{Phi}TSC2-/- vs control M{Phi} had basal increased mTORC1 and reduced mTORC2 activity. M{Phi}TSC2-/- were differentially responsive to stimulation by lipopoly-saccharide/IFN-{gamma} or IL-4 in vitro, and all disparities were prevented by rapamycin confirming the model. In vivo, M{Phi} TSC2-/- mice were strongly protected against I/R injury, with minimal change in ejection fraction, less LV dilation, hypertrophy, lung edema, or activation of stress/pro fibrotic genes. Mice pre-treated with anti-LY6G Ab to deplete neutrophils were still similarly protected, suggesting that the impact was primarily related to M{Phi}. M{Phi}TSC2-/- mice had less myocardial pro-inflammatory macrophages (CCR2+MHC-IIhi), LY6C+ monocytes, neutrophils, and CD8+ T cells 5 days post-I/R, and fewer CCR2+ but more CCR2- M{Phi} 2 weeks post I/R. Both M{Phi}TSC2-/- in vitro and in vivo post I/R phenotypes were converted to WT by rapamycin, supporting mTORC1 dependence. Lastly, synthesis of glycoprotein nonmetastatic melanoma protein B (GPNMB), a principally M{Phi} anti-inflammatory secreted protein protective against myocardial infarction was enhanced in M{Phi}TSC2-/- macrophages and hearts following I/R in an mTORC1 dependent manner. Conclusion: Constitutive macrophage-specific mTORC1 activation via TSC2 deletion reduces pro-inflammatory cell infiltration, increases GPNMB protein expression and preserves heart function following I/R injury. Rapamycin eliminates these effects. These results identify a cardioprotective mTORC1-GPNMB signaling nexus in M{Phi} in vivo.