Long read and short read whole genome sequencing are equivalent for genomic characterisation of bacteriophage: considerations for high throughput analysis
Long read and short read whole genome sequencing are equivalent for genomic characterisation of bacteriophage: considerations for high throughput analysis
Carr, P. G.; Iszatt, J. J.; Hedges, M. G.; Mantjani, L.; Vaitekenas, A.; Stick, S. M.; Kicic, A.; Montgomery, S. T.; Phage WA,
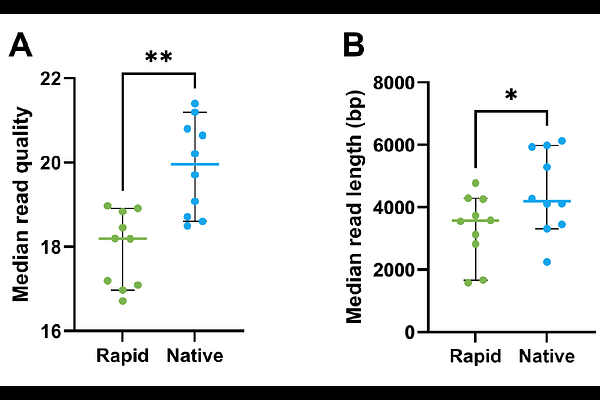
AbstractBackground: Antimicrobial resistance (AMR) is a global health crisis, necessitating alternative antibacterial strategies. Bacteriophages (phages) offer a promising solution, and their use as a therapeutic agent relies on stringent bioinformatic characterisation using whole genome sequencing (WGS) technologies. However, phages are highly diverse, with no clear consensus on best practices concerning phage DNA extraction or sequencing platform. Efficient and repeatable DNA extraction, sequencing, and bioinformatics processes are critical for safety assessments but remain poorly defined. Additionally, the impact of sequencing platform choice and DNA extraction methods on downstream genomic analyses is not well understood. Methods: We evaluated multiple DNA extraction, library preparation, and sequencing approaches using a diverse collection of Pseudomonas phages from the PhageWA biobank. Column-based and precipitation-based DNA extraction methods were compared for DNA yield and recovery efficiency. Genome sequencing was performed using short-read (Illumina) and long-read (Oxford Nanopore Technologies) platforms, incorporating multiple library preparation kits and Nanopore basecalling models. Assemblies were assessed for completeness, quality, and sequence concordance using standardised bioinformatics pipelines, with hybrid Illumina-Nanopore assemblies used as references for comparison. Results: DNA extraction efficiency varied substantially between protocols, with the Puregene precipitation-based method yielding significantly higher DNA recovery than column-based approaches when normalised to phage titre. Illumina sequencing consistently generated complete genome assemblies, although assembly fragmentation was observed for several jumbo phages when using the SeqWell ExpressPlex 2.0 library preparation method. For Nanopore sequencing, ligation-based native barcoding libraries produced longer reads than rapid barcoding libraries, while selection of the Dorado v5.0.0 basecalling model significantly improved read quality. Genome assembly success was dependent on phage genus; native Nanopore sequencing failed to assemble several Pbunavirus genomes, likely due to modified DNA bases, but an amplification-based library preparation successfully resolved these genomes. Across successfully assembled samples, Illumina and Nanopore platforms produced highly concordant genomes with comparable completeness scores, and hybrid polishing identified only minor sequence differences. Conclusions: DNA extraction methodology, sequencing chemistry, and basecalling model selection significantly influence phage WGS outcomes. Precipitation-based DNA extraction improved DNA recovery, while both Illumina and Nanopore sequencing generated high-quality phage genomes suitable for therapeutic characterisation. Nanopore sequencing provided assemblies comparable to Illumina with minimal benefit from hybrid polishing, supporting its routine use for phage genomics. These findings provide practical guidance for phage genome characterisation workflows and contribute to the development of standardised, regulatory-grade approaches for therapeutic phage assessment.