Molecular surveillance of multiplicity of infection, haplotype frequencies, and prevalence in infectious diseases
Molecular surveillance of multiplicity of infection, haplotype frequencies, and prevalence in infectious diseases
Tsoungui Obama, H. C. J.; Schneider, K. A.
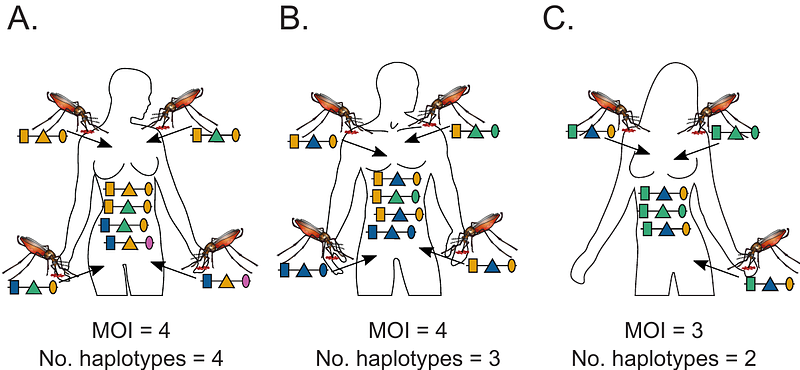
AbstractBackground: The presence of multiple different pathogen variants within the same infection, referred to as multiplicity of infection (MOI), confounds molecular disease surveillance in diseases such as malaria. Specifically, if molecular/genetic assays yield unphased data, MOI causes ambiguity concerning pathogen haplotypes. Hence, statistical models are required to infer haplotype frequencies and MOI from ambiguous data. Such methods must apply to a general genetic architecture, when aiming to condition secondary analyses, e.g., population genetic measures such as heterozygosity or linkage disequilibrium, on the background of variants of interest, e.g., drug-resistance associated haplotypes. Methods and Findings: Here, a statistical method to estimate MOI and pathogen haplotype frequencies, assuming a general genetic architecture, is introduced. The statistical model is formulated and the relation between haplotype frequency, prevalence and MOI is explained. Because no closed solution exists for the maximum-likelihood estimate, the expectation-maximization (EM) algorithm is used to derive the maximum-likelihood estimate. The asymptotic variance of the estimator (inverse Fisher information) is derived. This yields a lower bound for the variance of the estimated model parameters (Cramer-Rao lower bound; CRLB). By numerical simulations, it is shown that the bias of the estimator decrease with sample size, and that its covariance is well approximated by the inverse Fisher information, suggesting that the estimator is asymptotically unbiased and efficient. Application of the method is exemplified by analyzing an empirical dataset from Cameroon concerning anti-malarial drug resistance. It is shown how the method can be utilized to derive population genetic measures associated with haplotypes of interest. Conclusion: The proposed method has desirable statistical properties and is adequate for handling molecular consisting of moderate number of multiallelic molecular markers. The EM-algorithm provides a stable iteration to numerically calculate the maximum-likelihood estimates. An efficient implementation of the algorithm alongside a detailed documentation is provided as supplementary material.