A Pilot Study on the Urinary Microbiome Composition and Diversity in Clinical UTI Samples: A 16S rRNA Analysis
A Pilot Study on the Urinary Microbiome Composition and Diversity in Clinical UTI Samples: A 16S rRNA Analysis
Almamoori, A. A.; Farhan, M. H.; Al-Khafaji, N.; Al_Rahhal, A.
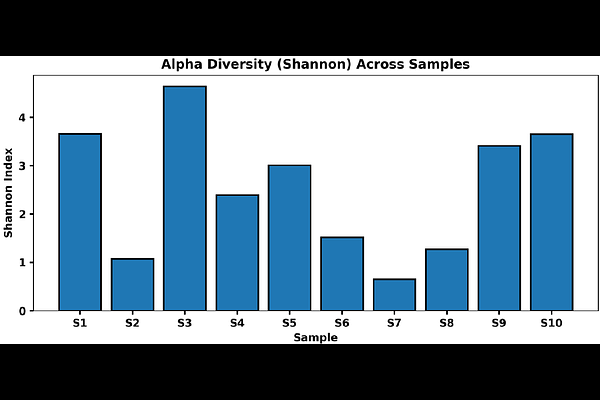
AbstractThis pilot study assessed the composition and diversity of the urinary microbiome from clinically confirmed UTI samples using 16S rRNA sequencing, whilst also exploring inter-individual variability of microbial community structure. We examined ten urine samples from patients with culture-positive UTIs. Demographic and clinical metadata, including age, sex, body mass index (BMI), diabetes status and recent antibiotic exposure was recorded per sample. Metagenomic DNA was extracted from microbial samples and sequenced to generate genus-level taxonomic profiling through 16S rRNA gene sequencing. Relative abundance tables were generated for each of the samples to identify dominant bacterial genera within each sample and summarize cohort level microbial patterns. To evaluate within-sample richness and evenness, alpha diversity indices (Shannon, Simpson, observed features and Chao1) were computed; beta diversity was measured using Bray-Curtis dissimilarity with principal coordinates analysis (PCoA) for graphical representation. The study's findings revealed the sex and moderate clinical diversity of the study sample; all samples were confirmed as having been taken from a UTI patient and exhibited a wide level of heterogeneity regarding the microbial composition of each urine sample. Overall, Pseudomonas was the dominant genus present, however, specific samples had approximately 50% of their microbiomes composed of Klebsiella, Proteus, and Escherichia species as well as approximately 25% of their total microbes were made up of Burkholderia spp., which are closely related to the genus of interest used during the course of this study. The observed alpha diversity of each sample displayed considerable variation for the included samples with a continuum of samples ranging from a single dominant microbe to a highly diverse mixed population producing a highly diverse polymicrobial population/bacterial composition, with some ratios of individual taxa to collective taxa of many samples repeatedly illustrating the exact nature of the specimen. Furthermore, a significant degree of Beta diversity was found between the patients, providing compelling evidence of identifiable differences among urinary microbiomes between patients with UTI. This pilot project provides a clear indication of the diversity and overall heterogeneity of urinary microbiota found in the UTI patients studied. In addition, the results of this study support the notion that the ecological complexities present within a urinary microbiome cannot necessarily be established through conventional culture methods, and that combined with molecular techniques such as 16S rRNA sequencing of bacterial DNA could be used to quantify and characterize the ecologic condition of urinary microbiota separate from the traditional high prevalence of identifiable uropathogens.