Bi-allelic mutations in KCTD11 cause a new form of autosomal recessive intermediate Charcot-Marie-Tooth disease
Bi-allelic mutations in KCTD11 cause a new form of autosomal recessive intermediate Charcot-Marie-Tooth disease
Gadacha, J.; Haidar, Z.; Roeckel-Trevisiol, N.; Pauset, A.; Castro, C.; Provost, C.; Hamze, Z.; Humbert, C.; Bertaux, K.; Lenfant, N.; Masingue, M.; de Becdelievre, A.; Konyukh, M.; Bonello, N.; Lia, A.-S.; Delmont, E.; Bertini, A.; Quartesan, I.; Facchini, S.; Cortese, A.; Reilly, M.; Houlden, H.; Pareyson, D.; Pisciotta, C.; Attarian, S.; Urtizberea, A.; Megarbane, A.; Jabbour, R.; Bernard-Marissal, N.; Delague, V.
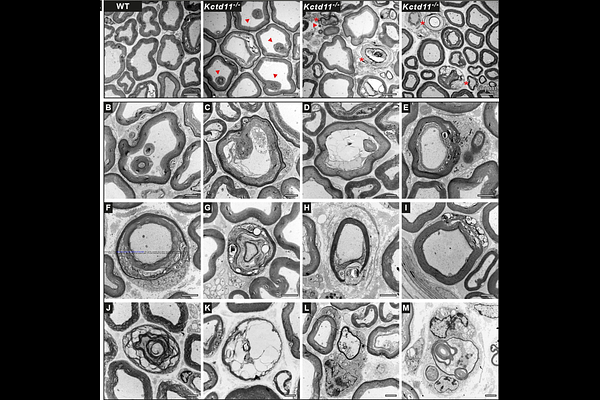
AbstractCharcot-Marie-Tooth disease (CMT) is the most common inherited neuromuscular disorder, characterized by progressive, length-dependent degeneration of peripheral nerves, resulting in distal muscle atrophy and weakness, foot and hand deformities, and sensory deficits. The disease is clinically and genetically heterogeneous, with over 125 disease-causing genes identified to date. Here, genetic studies in ten patients from 5 unrelated families of diverse ethnic background, led to the identification of KCTD11 as a novel CMT gene, responsible for a new autosomal recessive intermediate CMT subtype, RI-CMTE. The variants identified are loss of function. KCTD11 encodes KCTD11/REN, a protein of yet unknown function in the Peripheral Nervous System, known to regulate HDAC1, beta-catenin, and mTORC1, key regulators of myelination and neuronal differentiation in the PNS. To explore the role of KCTD11 in the PNS, we used a constitutive Kctd11-/- mouse model and the derived in vitro myelin model of sensory neuron and Schwann cell co-culture (DRGN/SC), to mimic the loss-of-function induced by patient mutations. We first demonstrate that the loss of KCTD11 is due to enhanced degradation of the mutated protein via autophagy. Both in vitro and in vivo, we demonstrate abnormal myelination in vivo and altered myelination dynamics in vitro. These defects were associated with dysregulation of the expression of key transcription factors in Schwann cells, such as Egr2 and Sox10, along with other myelin-related genes, as revealed by mRNA-sequencing data. Regarding pathophysiological mechanisms, we identified dysregulation of HDAC1 expression, as well as alterations in the Wnt/beta-catenin, Sonic Hedgehog and Hippo/YAP signaling pathways. The deregulation of these pathways seem to converge to altered autophagy and altered balance between proliferation, differentiation and apoptosis, at least in Schwann cells. These mechanisms remain to be explored in axons from PNS neurons. Altogether, our results identify KCTD11 as a novel gene defective in autosomal recessive intermediate RI-CMTE and highlight the key role of KCTD11 in maintaining myelin homeostasis through regulation of HDAC1 and phosphorylated beta-catenin levels, thereby preventing late-onset myelin abnormalities and degradation.