Somatic DNA methylation heterogeneity predicts extreme transgenerational epimutation hotspots in Arabidopsis
Somatic DNA methylation heterogeneity predicts extreme transgenerational epimutation hotspots in Arabidopsis
Vo, B. T.; Wolf, P.; Kim, J.; Zhang, Z.; Ramirez, V.; Poppenberger, B.; Schneitz, K.; Becker, C.; List, M.; Johannes, F.
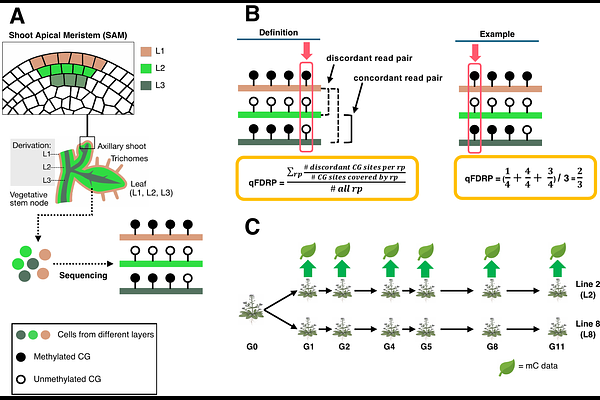
AbstractSpontaneous epimutations are stochastic and heritable changes in cytosine methylation that arise independently of DNA sequence alterations. In plants, they occur predominantly at CG sites, accumulate across generations at high and clock-like rates, and contribute substantially to constitutive epigenetic diversity. Although these features are well established, the developmental origin of spontaneous epimutations remains poorly understood. One model proposes that they arise during somatic growth in different cell layers of the shoot apical meristem, where lineage bottlenecks enable nascent epimutations to clonally propagate into developing organs. Such layer-specific dynamics are expected to generate spatial mosaics of methylation states within tissues that manifest as heterogeneity in bulk bisulfite sequencing data. Here, we quantified genome-wide CG methylation heterogeneity in single Arabidopsis leaves using read-level methylation discordance metrics adopted from cancer epigenomics. We identified thousands of highly heterogeneous loci and show that they overlap extreme transgenerational epimutation hotspots. Analysis of multiple leaves from the same individuals further revealed that methylation divergence at these loci scales with developmental distance and recapitulates the branching architecture of the shoot. Together, these results support a meristematic origin of spontaneous epimutations and highlight a shared susceptibility to methylation-maintenance errors in both developmental and transgenerational contexts.