Collectin-11 regulates osteoclastogenesis and bone maintenance via a complement-dependent mechanism.
Collectin-11 regulates osteoclastogenesis and bone maintenance via a complement-dependent mechanism.
Howard, M.; Farrar, C. A.; Nauser, C.; Jeon, Y.; Polycarpou, A.; Smolarek, D.; Greenlaw, R.; Garred, P.; Alexandra, D.; Mukhopadhyay, S.; Sacks, S.
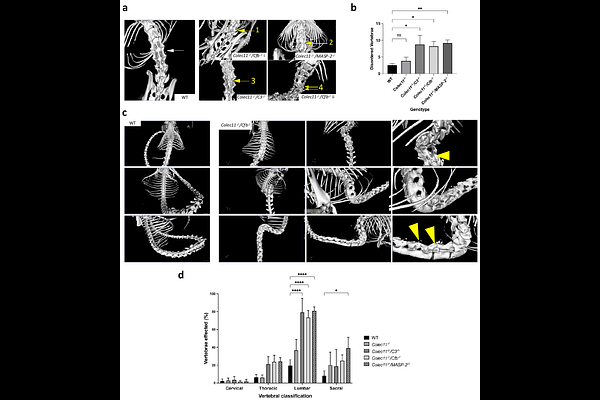
AbstractThe human developmental disorder 3MC syndrome is characterized by skeletal deformities associated with a deficiency of the pattern recognition molecule collectin-11 (CL-11), yet the underlying molecular and cellular mechanisms remain unclear. Here, we demonstrate that CL-11 deletion alone does not cause bone abnormalities in mice; however, combined deficiencies involving CL-11 and complement components MASP-2 (lectin pathway), CFB, or C3 (alternative amplification pathway) lead to significant vertebral bone loss and spinal curvature by 12 weeks of age. Ex vivo osteoclast (OCL) differentiation from bone marrow-derived cells of these double-knockout (DKO) mice was markedly impaired, but differentiation capacity was substantially restored by supplementation with CL-11. Furthermore, CL-11 and membrane attack complex (C5b-9) deposition were co-localized to OCLs and their precursors in normal bone tissues from embryonic stages to adulthood. These findings identify CL-11 as a critical osteoclastogenesis and bone maintenance regulator in conjunction with complement system-mediated signalling pathways and highlight CL-11 as a potential therapeutic target in diseases involving dysregulated osteoclast function and bone remodelling.