Tau-induced mitochondrial reverse electron transport drives neurodegeneration
Tau-induced mitochondrial reverse electron transport drives neurodegeneration
Li, W.; Rimal, S.; Bhurtel, S.; Yeung, L.; Lu, B.; Grinberg, L. T.; Spina, S.; Cobos, I.; Seeley, W. W.; Guo, S.; Lu, B.
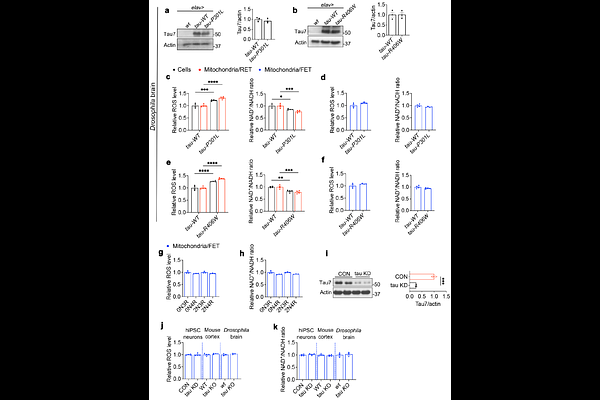
AbstractHyperphosphorylation and aggregation of the microtubule-associated protein tau are recognized as pathological hallmarks of tauopathies; however, the biological activity of tau that drives its pathophysiological effects remains poorly understood. Mitochondrial dysfunction is a common feature of tauopathies. Despite this, the mechanistic link between tau abnormalities and mitochondrial dysfunction, as well as its relationship to the physiological function of tau, remains unclear. Here, we demonstrate that tau regulates mitochondrial reverse electron transport (RET), which produces excess ROS, reduces the NAD+/NADH ratio, and is activated by aging or stress. In flies, mice, and human induced pluripotent stem cells (hiPSC)-derived neurons, tau depletion eliminates stress-induced RET and confers significant stress resistance. Mechanistically, tau enters mitochondria and directly interacts with the mitochondrial complex I (C-I) subunit NDUFS3, enhancing RET activation in a phosphorylation-dependent manner that correlates with tau pathogenicity. Elevated RET further drives tau hyperphosphorylation, establishing a self-perpetuating pathological loop. Blocking tau entry into mitochondria or disrupting tau/NDUFS3 interaction reduces tau-induced RET. Genetic or pharmacological inhibition of RET protects against tau-induced neurodegeneration across species. RET regulation represents a previously unrecognized normal function of tau that becomes pathological in disease, providing a therapeutic target for conditions characterized by tau abnormalities and mitochondrial dysfunction.