KMT2C and KMT2D regulate skeletal development through stage-specific epigenetic control of chondrogenesis
KMT2C and KMT2D regulate skeletal development through stage-specific epigenetic control of chondrogenesis
Quickstad, G.; Bikas, D. V.; Vardabasso, S.; Shpargel, K. B.
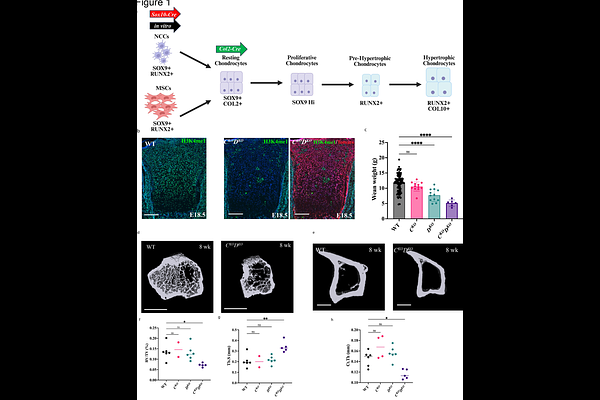
AbstractMany craniofacial disorders linked to mutations in enhancer-associated chromatin-modifying enzymes, including Kabuki syndrome (KS), present with a wide range of skeletal abnormalities. KS is a craniofacial development disorder characterized by mutations in KMT2D, a histone H3 lysine 4 (H3K4) methyltransferase. The KMT2D cellular origins and molecular pathways leading to skeletal deficits in KS are not well characterized. We previously demonstrated that KMT2D deletion in mouse cranial neural crest cells (cNCCs), the multipotent stem cells that contribute to many craniofacial structures, specifically impairs chondrocyte hypertrophic differentiation. Although we observed defects in cNCC endochondral ossification, broader skeletal function of KMT2D is largely unknown and its functional redundancy in skeletal development with its paralog, KMT2C, is understudied. We now find that KMT2D mutation within chondrocyte lineages results in postnatal growth defects and impaired skeletal bone development. We demonstrate redundancy of KMT2C in regulating endochondral ossification as mice lacking both methylases within chondrocyte lineages demonstrate severe deficits in bone formation. These methylases regulate change in chondrocyte state as hypertrophic differentiation is impaired with loss of both KMT2C and KMT2D. When both methylases are deleted in stem cell lineages, KMT2C/KMT2D double mutant cNCCs are unable to properly induce chondrocyte factors downstream of SOX9 activation. At the molecular level, there is a loss of H3K4me1 at de novo chondrocyte enhancers essential for skeletal development and an associated reduction in the efficiency of acetylation with KMT2C/D deficiency, suggesting partial dependency on KMT2C/D presence for appropriate deposition of H3K27ac. We propose a role for KMT2C and KMT2D in facilitating proper activation of genes downstream of SOX9 and RUNX2 transcription factors. Thus, we delineate a regulatory cascade orchestrated by chromatin-level modifications wherein KMT2C/D activity ensures commitment of chondrocyte lineages via enhancer priming. This work not only expands the functional repertoire of COMPASS family members but also defines a chromatin-based mechanism for vertebrate skeletal development, offering insights into the epigenetic basis of skeletal dysplasia and related congenital disorders.