Panmap: Scalable phylogeny-guided alignment, genotyping, and placement on pangenomes
Panmap: Scalable phylogeny-guided alignment, genotyping, and placement on pangenomes
Kramer, A. M.; Zhang, A.; Ayala, N.; de Sanctis, B.; Karim, L. M.; Hinrichs, A. S.; Walia, S.; Turakhia, Y.; Corbett-Detig, R.
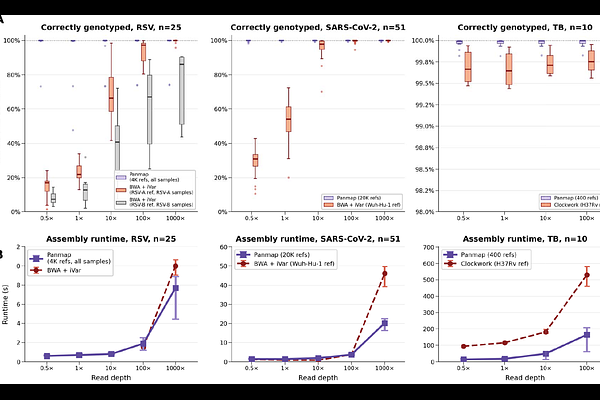
AbstractPangenomes capture population-level variation but remain computationally challenging at scale. We present Panmap, a tool that leverages evolutionary structure to place, align, and genotype sequencing reads against mutation-annotated pangenomes containing up to millions of genomes. Panmap introduces a phylogenetically compressed k-mer index that stores only sequence differences along branches, enabling efficient comparison of reads to both sampled genomes and inferred ancestors. This approach reduces index size by up to 600-fold and construction time by over three orders of magnitude relative to existing tools. Panmap places a 100x coverage SARS-CoV-2 sample onto 20,000 genomes in 0.4 seconds and onto 8 million genomes in under two minutes. Furthermore, it enables accurate haplotype identification and abundance estimation in metagenomic samples and sensitive placement of ancient environmental DNA without prior alignment. Our approach makes large-scale pangenomes directly amenable to read mapping, genome assembly, alignment-free phylogenetic placement, and metagenomic analysis.