Evaluating the use of non-linear models in data-driven rescoring of peptide-spectrum matches
Evaluating the use of non-linear models in data-driven rescoring of peptide-spectrum matches
Nameni, A.; Declercq, A.; Gabriels, R.; Degroeve, S.; Martens, L.; Bouwmeester, R.
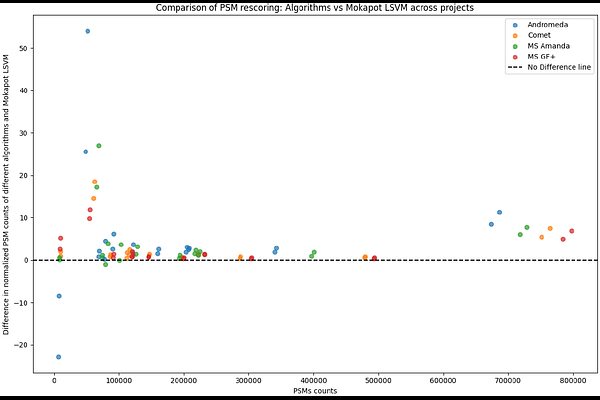
AbstractIn mass spectrometry (MS)-based proteomics, computational tools match acquired tandem MS spectra to peptides from a sequence database. Machine learning increasingly supports this task through peptide-spectrum match (PSM) rescoring, in which a classifier, typically a linear semi-supervised model, refines the initial matching score. However, Mokapot allows the user to choose among different machine learning algorithms of increasing complexity, from the default linear support vector machine (LSVM) to random forest and XGBoost. Here, we use an entrapment approach to assess the effect of this increasing complexity on PSM identification and the accuracy of the estimated false discovery rate (FDR). We show that, while more complex models increase the number of identified PSMs at a fixed FDR threshold, this gain reflects a bias towards random matches from the target proteome database rather than genuine identifications. Indeed, for the most complex model, the entrapment FDR reaches 6.3% instead of the estimated 1% decoy FDR. This bias thus yields overly optimistic FDR estimates, indicating that model complexity in PSM rescoring must be carefully balanced against this overfitting risk.