Integrative genomics of Plasmodium knowlesi reveals parasite-intrinsic regulators of severe human malaria
Integrative genomics of Plasmodium knowlesi reveals parasite-intrinsic regulators of severe human malaria
Westaway, J. A.; Benavente, E. D.; Kucharski, M.; Nayak, S.; Huy, D. T.; Auburn, S.; William, T.; Rajahram, G. S.; Piera, K. A.; Trimarsanto, H.; Hoon, K. S.; Bourke, C.; Jantim, A.; Manah, A. M.; Gotulis, W.; Moon, R. W.; Amato, R.; Barber, B. E.; Drakeley, C.; Anstey, N. M.; Field, M. A.; Bozdech, Z.; Grigg, M. J.
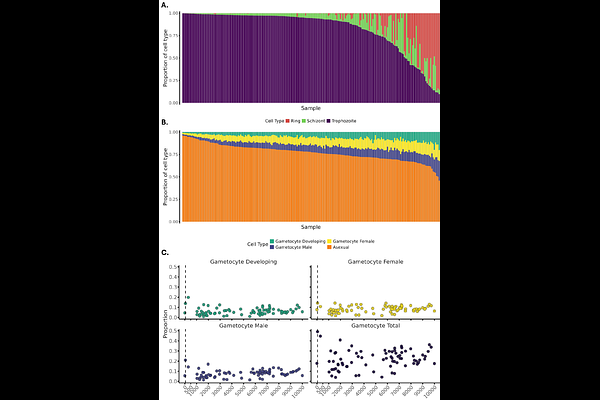
AbstractTo dissect parasite determinants of clinical disease severity in Plasmodium knowlesi, we performed the first integrative genome-wide association (GWAS), transcriptomic, and expression quantitative trait locus (eQTL) analysis from clinical malaria isolates. We identify distinct parasite programs associated with WHO-defined severe disease, characterized by transcriptional activation of stress-response and host-interaction pathways. In contrast, invasion-linked programs were more strongly associated with parasite burden, while immune-evasion pathways showed overlapping but distinct associations with both burden and severity. GWAS and eQTL analyses revealed genetic regulation of transcriptional states, including immune-evasion variant antigen families (SICAvar and kir) and chromatin-associated regulators. Mature gametocyte transcripts were detectable across infections, including those with low parasitaemia, indicating that transmission-stage expression occurs independently of both clinical severity and parasite density. Together, these findings show that severe P. knowlesi malaria is associated with genetically regulated parasite transcriptional programs that are not fully explained by parasite burden.