Sequencing depth overcomes extraction bias: repurposing human WGS data for salivary microbiome profiling
Sequencing depth overcomes extraction bias: repurposing human WGS data for salivary microbiome profiling
Velo-Suarez, L.; Herzig, A. F.; Bocher, O.; Le Folgoc, G.; Le Roux, L.; Delmas, C.; Zins, M.; Deleuze, J.-F.; Hery-Arnaud, G.; Genin, E.
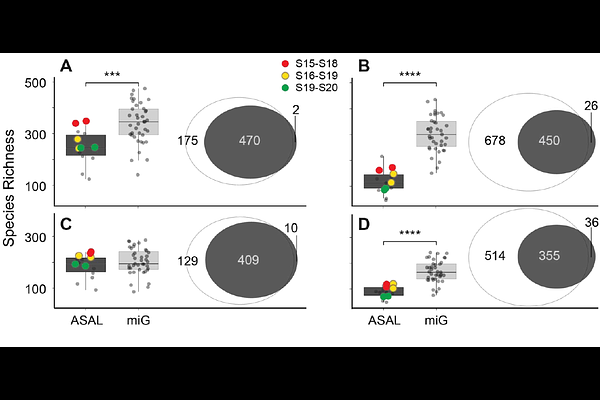
AbstractLarge-scale human genomic projects have generated whole-genome sequencing (WGS) data from hundreds of thousands of individuals, primarily to study host genetic variation. When saliva is the DNA source, the resulting datasets also contain microbial reads that are routinely discarded. Here, we investigate whether these host-centric WGS workflows can yield reliable microbiome profiles, effectively doubling the research value of existing data without additional sampling. We compared non-human reads from 39 deeply sequenced saliva samples from the GAZEL cohort (miG dataset; median ~43 million reads/sample) with 14 samples processed with microbiome-optimized extraction (ASAL; median ~4.3 million reads/sample), using two complementary classifiers: meteor, a coverage-based mapper against a curated saliva-specific database, and sylph, a k-mer classifier against the Genome Taxonomy Database (GTDB). Despite the absence of microbial lysis optimization, miG samples showed up to 3-fold higher species richness, ~10-fold greater sequencing depth, and significantly lower inter-sample variability (PERMANOVA R{superscript 2} = 0.10, p = 0.001; BETADISPER p = 0.0036). Rarefaction to 10? reads eliminated most compositional differences, demonstrating that sequencing depth is the primary driver of community stability. Only ~2% of detected taxa (12 of 592) showed extraction-related differences. The two classifiers exhibited fundamentally different depth-sensitivity profiles, with sylph retaining systematic detection asymmetries even after depth normalization, highlighting that classifier choice introduces biases that affect cross-study comparisons. These results show that biobank WGS data from saliva can be repurposed for robust, population-scale oral microbiome analyses, enabling simultaneous investigation of host genomic variation and the microbiome from the same archived samples.