Histone Modification Metapeaks are Epigenetic Landmarks Predictive of Cell State
Histone Modification Metapeaks are Epigenetic Landmarks Predictive of Cell State
Tanner, R. M.; Perkins, T. J.
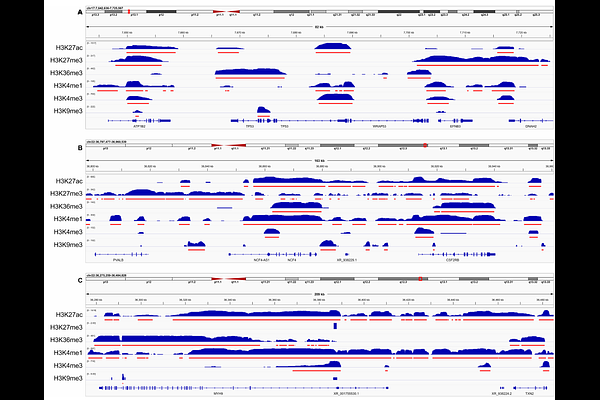
AbstractHistone modifications are a key component of the epigenetic state of a cell, and they vary widely across different cell and tissue types, conditions, and disease states. Indeed, the majority of the genome is enriched with one histone mark or another across the thousands of cellular conditions that have been studied to date. Here, we use the largest-to-date collection of histone modification ChIP-seq datasets to identify the most important sites of histone modifications genome-wide. Collected and uniformly reprocessed by the International Human Epigenome Consortium, this data includes 5339 datasets enriched at nearly one billion total peaks across 59 different major cell or tissue types and in healthy and disease conditions, for six different histone marks. We propose FindMetapeaks, a new approach to identifying histone mark metapeaks, which are genomic regions with enrichment of a mark across many samples. We show that many of these epigenetic metapeaks are strongly indicative of cell and tissue type, or are associated with other sample characteristics, and highlight key regulatory regions of the genome. However, we also show that many metapeaks contain redundant information, and that parsimonious subsets of metapeaks can be selected by machine learning to predict cell state. Our histone mark metapeak atlas provides a concise set of regions for interpreting the epigenome.