Residue burial encodes a protein's fold
Residue burial encodes a protein's fold
Grigas, A. T.; Sumner, J.; O'Hern, C. S.
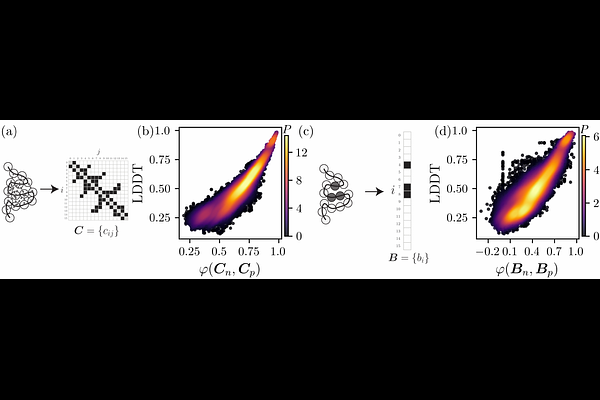
AbstractProtein structure is controlled by a high-dimensional energy landscape, which is a function of all of the atomic coordinates of the protein. Can this landscape be accurately described by a low-dimensional representation? We find that residue core identity, a binary N-dimensional encoding indicating whether each of the N amino acids in a protein is buried in the core or not, can predict the protein's backbone conformation more efficiently than all other representations that we tested. Core identity is 4 times more efficient than previous estimates of the bits per residue needed to encode a protein's native fold, 2 times more efficient than the C contact map, and 1.5 times more efficient than the machine-learned embeddings from FoldSeek's 3Di. Even when the folded structure is unavailable, predicting each residue's burial from sequence yields a more accurate estimate of fold quality than predicting pairwise contacts from the same sequence information. Thus, this work emphasizes that the problem of determining a protein's native fold can be re-framed as predicting each residue's core identity.