Ubiquitin ligase CHFR impairs Tie2 signaling via K48-linked ubiquitylation and degradation of Akt1 in endothelial cells
Ubiquitin ligase CHFR impairs Tie2 signaling via K48-linked ubiquitylation and degradation of Akt1 in endothelial cells
Tiruppathi, C.
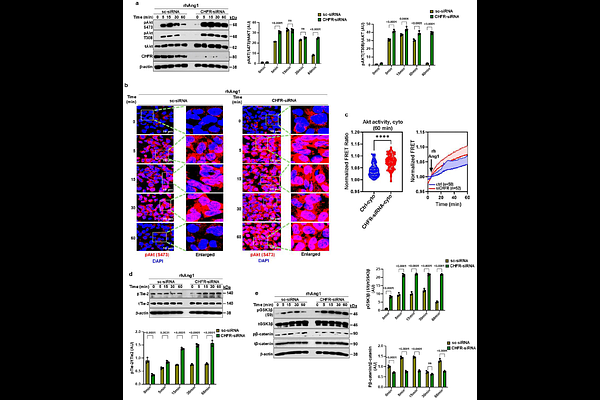
AbstractVascular endothelial (VE)-cadherin is essential for maintaining endothelial junctional barrier integrity. The Angiopoietin-1 (Ang-1)/Tie2 axis induced Akt1 activation is crucial for maintaining endothelial junctional barrier by inhibiting FoxO1 and suppressing expression of Angiopoietin-2 (Ang-2), a Tie2 antagonist. Systemic inflammatory conditions such as sepsis, Akt1 expression is reduced, whereas FoxO1-dependent Ang-2 expression is increased, resulting in endothelial barrier dysfunction. We previously showed that the TLR4/FoxO1 axis induces the ubiquitin E3 ligase CHFR, which promotes endothelial barrier disruption by targeting VE-cadherin for ubiquitylation and degradation. However, little is known about Akt1 expression during vascular inflammation. Here, we identified FoxO1-dependent CHFR expression as a key mechanism driving K48-linked polyubiquitylation and proteasomal degradation of Akt1 in endothelial cells (EC). LPS-induced K48-linked ubiquitylation of Akt1 was prevented in CHFR-depleted human EC and in endothelial-specific Chfr knockout (Chfr{Delta}EC) mice. Accordingly, CHFR depletion increased Akt1 and VE-cadherin expression in both human lung EC and Chfr{Delta}EC mice. Chfr{Delta}EC mouse lungs also exhibited elevated Ang-1 and Tie2 expression, and Ang-1 stimulation induced sustained Akt1 phosphorylation in CHFR-deficient EC. Moreover, CHFR depletion prevented LPS-induced expression of FoxO1 and Ang-2 in EC. Mechanistically, CHFR interacted with phosphorylated Akt1 and mediated its ubiquitylation at lysine residues K30, K39, K154, and K268. Expression of a ubiquitylation-deficient Akt1 mutant prevented LPS-induced VE-cadherin degradation and vascular injury. Collectively, these findings identify CHFR as a critical regulator of endothelial inflammatory responses by controlling Akt1 stability and VE-cadherin expression during inflammation.