Scalable Inference and Identifiability of Kinetic Parameters for Transcriptional Bursting from Single Cell Data
Scalable Inference and Identifiability of Kinetic Parameters for Transcriptional Bursting from Single Cell Data
Gu, J.; Laszik, N.; Miles, C. E.; Downing, T. L.; Allard, J.; Read, E. L.
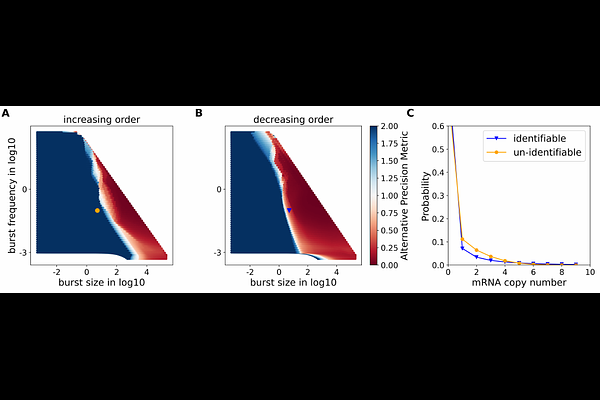
AbstractStochastic gene expression and cell-to-cell heterogeneity have attracted increased interest in recent years, enabled by advances in single-cell measurement technologies. These studies are also increasingly complemented by quantitative biophysical modeling, often using the framework of stochastic biochemical kinetic models. However, inferring parameters for such models (that is, the kinetic rates of biochemical reactions) remains a technical and computational challenge, particularly doing so in a manner that can leverage high-throughput single-cell sequencing data. In this work, we develop a Chemical Master Equation reference library-based computational pipeline to infer kinetic parameters describing noisy mRNA distributions from single cell RNA sequencing data, using the commonly applied stochastic telegraph model. Our pipeline also serves as a tool for comprehensive analysis of parameter identifiability, in both a priori (studying model properties in the absence of data) and a posteriori (in the context of a particular dataset) use cases. The pipeline can perform both of these tasks, i.e. inference and identifiability analysis, in an efficient and scalable manner, and also serves to disentangle contributions to uncertainty in inferred parameters from experimental noise versus structural properties of the model. We found that for the telegraph model, the majority of the parameter space is not practically identifiable from single cell RNA sequencing data, and low experimental capture rates worsen the identifiability. Our methodological framework could be extended to other data types in the fitting of small biochemical network models.