Accurate estimation of canine inbreeding using ultra low-coverage whole genomesequencing
Accurate estimation of canine inbreeding using ultra low-coverage whole genomesequencing
Pellegrini, M.; Kim, R.; Rubbi, L.; Kislik, G.; Smith, D.
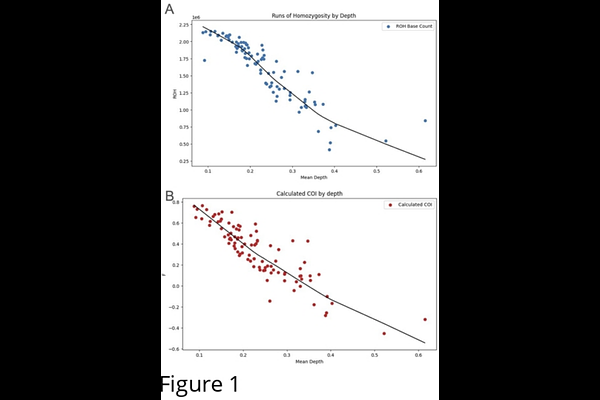
AbstractThe measurement of inbreeding has gained significance across diverse fields, including population and conservation genetics, agricultural genetics, breeding programs for animals and plants, and wildlife management. This is due to the fact that inbreeding leads to increased homozygosity and results in lower genetic diversity, rendering populations more vulnerable to environmental changes, diseases, and other stressors. High or mid-coverage whole genome sequencing (WGS) has been widely used for inbreeding estimation, but it is resource-intensive. We aimed to investigate the use of ultra low-coverage whole genome sequencing (ulcWGS) as a cost-effective alternative for inbreeding analysis. Domestic dogs were used for our study as their extensive breeding histories lead to populations with a wide range of inbreeding levels. We constructed a multi-breed reference panel from high-coverage WGS samples. Inbreeding in independent ulcWGS samples was then estimated using runs of homozygosity (RoH) and inbreeding coefficients (F). We modeled the relationship between these measures and sequencing depth using nonlinear regression, to generate inbreeding estimates relative to sequencing depth. Resulting relative RoH and F measurements were significantly correlated, with purebred dogs exhibiting more runs of homozygosity and higher inbreeding coefficients2 compared to mixed-breed dogs. Our findings demonstrate that ulcWGS can provide reliable and economical estimations of inbreeding, expanding accessibility to genetic monitoring.