Improving isoform-level eQTL and integrative genetic analyses of breast cancer risk with long-read RNA transcript assemblies
Improving isoform-level eQTL and integrative genetic analyses of breast cancer risk with long-read RNA transcript assemblies
Head, S. T.; Nemani, A.; Chang, Y.-H.; Harrison, T. A.; Bresnahan, S. T.; Rothstein, J. H.; Sieh, W.; Lindstroem, S.; Bhattacharya, A.
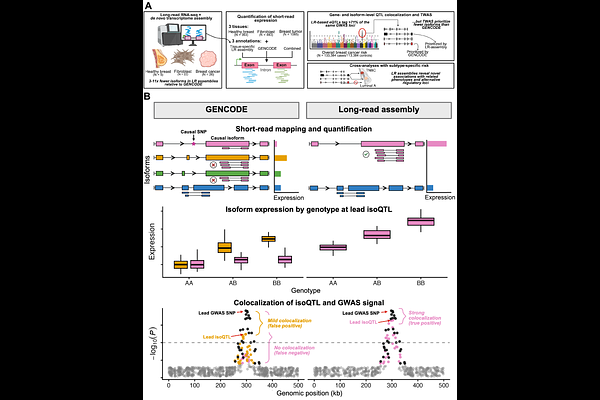
AbstractMost eQTL and TWAS analyses quantify expression using aggregate, tissue-agnostic transcript annotations and ignore isoform-level regulation, potentially obscuring or misattributing regulatory mechanisms. Here, we developed a framework leveraging publicly available long-read RNA-seq data to perform tissue-informed inference of genetic regulation and prioritize candidate causal isoforms for breast cancer risk. We quantified gene- and isoform-level expression in breast tumor (TCGA), non-cancerous mammary tissue, and cultured fibroblasts (GTEx) using three transcriptome annotations: standard GENCODE, tissue-specific long-read-derived assemblies, and combined annotations incorporating transcript-isoforms from both. While GENCODE cataloged over 250,000 pan-tissue isoforms, the tissue-specific long-read assemblies captured reduced sets of 74,717 isoforms in tumor, 48,057 in fibroblasts, and 22,941 in healthy breast. We performed eQTL mapping and fine-mapping, followed by colocalization with overall and subtype-specific breast cancer GWAS and isoform-level TWAS. While most eGenes were concordant across annotations, approximately 1/3 of lead cis-eQTLs for shared eGenes differed between long-read assemblies and GENCODE. Further, eIsoform discovery was highly annotation-specific. In healthy breast tissue, the gold standard tissue for building gene expression prediction models for TWAS of breast cancer, 46% of eIsoforms identified by the long-read annotation were unique to that annotation even though 93.7% of them are present in GENCODE. Despite combined annotations expanding the GENCODE catalog by only 0.6-7.6% depending on tissue source, 69% of unique significant isoform-trait associations were specific to a single annotation. Long-read-informed annotations uncovered regulatory associations entirely missed by GENCODE, including a candidate regulatory isoform at the MARK1 locus captured only in fibroblasts and a previously unannotated splice variant prioritized as the likely effector transcript at NUP107. These findings demonstrate that transcript annotation is not merely a technical consideration but critically defines the biological hypothesis space for regulatory mechanisms and shapes discovery. Incorporating tissue-resolved isoform annotations from long-read RNA-seq improves the specificity of regulatory inference and enhances identification of candidate causal isoforms at GWAS loci.