MolMAE: A Surface-Centric Multimodal Masked Autoencoder for Molecular Representation Learning
MolMAE: A Surface-Centric Multimodal Masked Autoencoder for Molecular Representation Learning
Li, J.
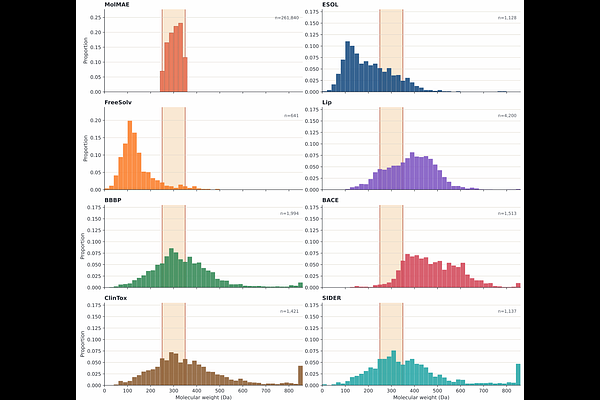
AbstractMolecular representation learning has become a central component of modern computational drug discovery. Existing molecular foundation models mainly rely on SMILES strings, two-dimensional molecular graphs, or three-dimensional atomic coordinates. However, many molecular properties are ultimately governed by the molecular surface, where intermolecular recognition, solvation, electrostatic complementarity, and ligand-protein interactions occur. In this work, we propose MolMAE, a surface-guided multimodal masked autoencoder for molecular representation learning. MolMAE takes molecular surface point clouds, three-dimensional molecular graphs, and SMILES-derived fragment and functional-group tokens as complementary input modalities, and learns a unified multimodal molecular embedding through functional-group-aligned masked autoencoding. During pretraining, chemically corresponding local regions are jointly masked across surface, graph, fragment, and functional-group views, forcing the model to reconstruct missing geometric, physicochemical, structural, and semantic information from the remaining context. While molecular surface reconstruction serves as the primary pretraining objective, graph-, fragment-, and functional-group-level reconstruction tasks provide complementary supervision that encourages the model to capture molecular topology, bonding patterns, stereochemistry, local chemical environments, and substructure organization. In addition to reconstructing surface geometry, MolMAE reconstructs surface-associated physicochemical fields, including electrostatic potential and Fukui-related descriptors, enabling the model to learn chemically meaningful surface representations. Pretrained on approximately 261K lead-like bioactive molecules, MolMAE achieves strong performance on the ESOL benchmark under scaffold splitting and competitive performance across multiple molecular property prediction tasks. These results suggest that molecular surface-guided pretraining can complement conventional graph-, sequence-, and atom-coordinate-based molecular representations, especially for property prediction tasks influenced by exposed surface geometry and surface-associated physicochemical patterns.