Mapping the Transcriptional Landscape of Drug Responses in Primary Human Cells Using High-Throughput DRUG-seq
Mapping the Transcriptional Landscape of Drug Responses in Primary Human Cells Using High-Throughput DRUG-seq
Baugh, L.; Vigneau, S.; Sridhar, S.; Boswell, S.; Pilitsis, G.; Bradley, J.; Allen, O.; Rand, L.; Riccardi, A.; Roach, K.; Lema, K.; Ergun, A.
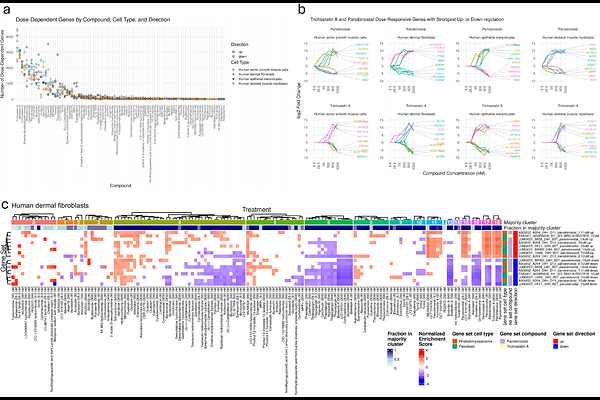
AbstractTo advance our understanding of drug action in physiologically-relevant systems, we developed a high-throughput transcriptomic atlas of compound responses in primary human cell types. Leveraging the scalable and cost-effective Digital RNA with the pertUrbation of Genes (DRUG-seq) assay, we profiled gene expression responses to 89 pharmacologically-active compounds across six concentrations in four distinct primary cell types: aortic smooth muscle cells (AoSMCs), skeletal muscle myoblasts (SkMMs), dermal fibroblasts, and melanocytes. Through rigorous quality control and normalization, we generated reproducible and cell type-resolved transcriptomic signatures, enabling the discovery of both shared and divergent regulatory programs. This dataset revealed core cellular responses, such as brefeldin A-mediated ER stress across all cell types, as well as lineage-specific effects, including dexamethasone-induced hypoxia signaling in AoSMCs, complex inflammatory responses linked to epithelial-to-mesenchymal transition pathways in SkMMs, TGF-{beta}-modulated states in fibroblasts, and dabrafenib-driven transcriptional shifts towards quiescence in melanocytes. By integrating systematic perturbations with primary models, this dataset serves as a resource for building systems-level models of drug response and mechanism. Ultimately, we aim to accelerate predictive pharmacology by enabling high-throughput data generation grounded in human biology and readily usable by artificial intelligence models.