Modeling acquired TKI resistance and effective combination therapeutic strategies in murine RET+ lung adenocarcinoma
Modeling acquired TKI resistance and effective combination therapeutic strategies in murine RET+ lung adenocarcinoma
Hinz, T. K.; Le, A. T.; Doan, T.; Ast, A.; Jaramillo, S.; Haines, S.; Navarro, A.; Patil, T.; Nemenoff, R. A.; Heasley, L. E.
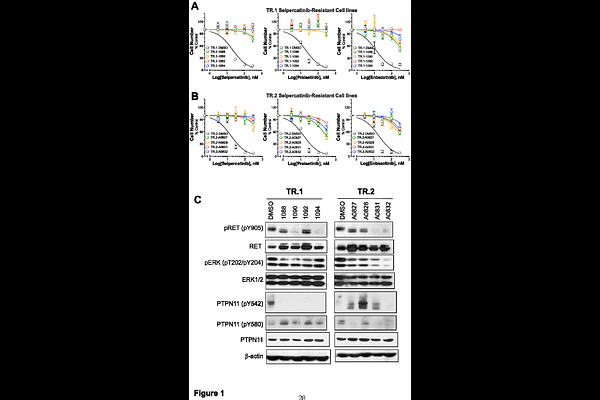
AbstractRET gene rearrangements yield oncogenic fusion proteins that drive a subset of lung adenocarcinomas (LUAD). The tyrosine kinase inhibitors (TKIs) selpercatinib and pralsetinib are approved therapies for RET+ lung cancers and have markedly improved clinical outcomes in these patients, but acquired resistance remains a hurdle to their durable management. Using a recently developed murine model of RET+ lung cancer driven by a Trim24-Ret fusion protein, two Trim24-Ret cell lines (TR.1 and TR.2) were established. Orthotopic lung tumors generated by transplantation of these cell lines initially respond to selpercatinib followed by prompt progression within ~3 weeks of initiating TKI treatment. Cell lines derived from the selpercatinib-resistant TR.1 and TR.2 tumors exhibited in vitro sensitivity to MET and ERBB-targeted TKIs, indicating acquired bypass signaling through these receptor tyrosine kinases. Moreover, the selpercatinib-resistant TR.1 and TR.2 cell lines exhibited increased sensitivity to MEK and PTPN11 inhibitors relative to the parental cell lines, indicating a greater dependence on MAPK pathway signaling. The TKI-resistant cell lines showed no evidence for MET gene amplification, but exhibited varied induction of multiple genes that function within MET and ERBB2:ERBB4 interaction networks including ligands (HGF, NRG1), adaptors (GAB1) and co-receptors (NRP1). Consistent with an important role for MET signaling in driving acquired selpercatinib resistance, mice bearing orthotopic lung tumors derived from TR.1 or TR.2 cells that had progressed on selpercatinib treatment underwent significant re-shrinkage upon co-treatment with the MET inhibitor, crizotinib, although progression re-occurred. By contrast, upfront treatment with selpercatinib and crizotinib in orthotopic tumors yielded complete elimination of 78% of TR.1 tumors and a prolonged duration of response in TR.2 tumors. The findings highlight the failings inherent in treating acquired resistance mechanisms at progression and the potential therapeutic impact of predicting and targeting dominant mechanisms of resistance prior to or early after initiating oncogene-targeting TKI treatment in RTK-driven LUAD.