Predicted mechanistic impacts of human protein missense variants
Predicted mechanistic impacts of human protein missense variants
Janes, J.; Muller, M.; Selvaraj, S.; Manoel, D.; Stephenson, J.; Goncalves, C.; Lafita, A.; Polacco, B.; Obernier, K.; Alasoo, K.; Lemos, M. C.; Krogan, N.; Martin, M.; Saraiva, L. R.; Burke, D.; Beltrao, P.
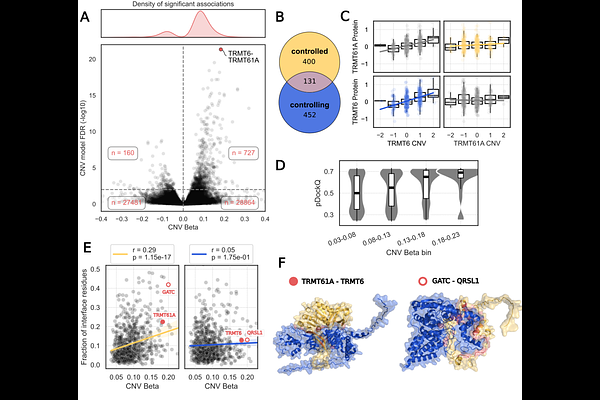
AbstractGenome sequencing efforts have led to the discovery of tens of millions of protein missense variants found in the human population with the majority of these having no annotated role and some likely contributing to trait variation and disease. Sequence-based artificial intelligence approaches have become highly accurate at predicting variants that are detrimental to the function of proteins but they do not inform on mechanisms of disruption. Here we combined sequence and structure-based methods to perform proteome-wide prediction of deleterious variants with information on their impact on protein stability, protein-protein interactions and small-molecule binding pockets. AlphaFold2 structures were used to predict approximately 100,000 small-molecule binding pockets and stability changes for over 200 million variants. To inform on protein-protein interfaces we used AlphaFold2 to predict structures for nearly 500,000 protein complexes. We illustrate the value of mechanism-aware variant effect predictions to study the relation between protein stability and abundance and the structural properties of interfaces underlying trans protein quantitative trait loci (pQTLs). We characterised the distribution of mechanistic impacts of protein variants found in patients and experimentally studied example disease linked variants in FGFR1.