PARP16 protects against cardiac hypertrophic response by ADP-ribosylation-dependent inhibition of NFAT transcription factor
PARP16 protects against cardiac hypertrophic response by ADP-ribosylation-dependent inhibition of NFAT transcription factor
Zarinfard, S.; Raghu, S.; Bangalore Prabhashankar, A.; Chowdhury, A.; Jayadevan, P.; Rajagopal, R.; Sharma, A.; Shrama, A.; MohanRao, P. S.; Nath, U.; Somasundaram, K.; Hottiger, M. O.; Sundaresan, N. R.
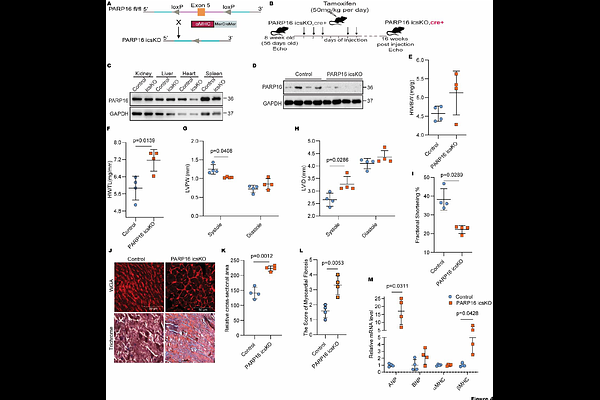
AbstractBACKGROUND: Mono-ADP ribosylation is a post-translational modification that regulates various cellular physiological processes, including cell cycle progression, genomic stability, transcription, and cellular protein turnover. PARP16 is an endoplasmic reticulum (ER)-localized mono-ADP-ribosyltransferase that has been shown to regulate the unfolded protein response and maintain ER homeostasis under stress conditions. Despite its established role in ER stress signaling, the functional significance of PARP16 in cardiac pathophysiology, particularly in cardiac hypertrophy and heart failure, remains poorly understood. In this study, we aim to investigate the role of PARP16 in cardiac hypertrophy and heart failure using in vitro and mouse model systems. METHODS: We analysed PARP16 expression in human heart failure samples as well as in heart failure-based mouse models. We evaluated gene expression by RT-PCR, immunoblotting, and confocal microscopy to understand the role of PARP16 in heart failure under phenylephrine- or isoproterenol-treated conditions. We also investigated the role of PARP16 in regulating cardiac function in genetically engineered mouse models, including whole-body PARP16 knockout, cardiac-specific PARP16 knockout, inducible cardiac-specific PARP16 knockout, and cardiac-specific PARP16 Transgenic mice. We performed echocardiography to assess cardiac function. We also used an in vitro primary cardiomyocyte system to knock down and overexpress PARP16. We performed RNA sequencing and mass spectrometry, followed by molecular docking, molecular dynamics simulation, immunoprecipitation, and luciferase assay to characterise the molecular mechanism by which PARP16 regulates cardiac function. RESULTS: Human heart failure samples showed reduced PARP16 expression. PARP16 expression was also significantly reduced in models of heart failure, including the hearts of isoproterenol-treated C57B/L6 mice and phenylephrine-treated primary cardiomyocytes. PARP16-deficient NRCMs showed signs of pathological remodelling. Whole-body, cardiac-specific, and inducible cardiac-specific PARP16 KO mice exhibited cardiac remodelling and dysfunction. In contrast, cardiac-specific PARP16-overexpressing mice were protected from iso-induced cardiac hypertrophy. Mechanistically, several hypertrophic signalling pathway genes are dysregulated in PARP16 knockout mouse hearts concomitant with upregulated NFAT1 transcriptional activity and nuclear translocation. PARP16 binds to and catalytically downregulates NFAT activity, thereby maintaining cardiac function. Mass spectrometry analysis showed that PARP16 is involved in ADP-ribosylation of NFAT1 at E398 and T533. Pharmacological inhibition of NFAT activation attenuates structural and functional abnormalities associated with PARP16 deficiency. CONCLUSIONS: PARP16 binds to and inhibits NFAT1 activity to regulate cardiac function in mice, and its downregulation may activate NFAT1 signalling, leading to hypertrophy. In this manner, PARP16 plays a critical role in cardiac hypertrophy and failure and may serve as a potential therapeutic target for the treatment of heart failure.