BxbI-mediated insertion of a 77kb human RET sensitive haplotype into the mouse genome to generate a humanized model of Hirschsprung disease
BxbI-mediated insertion of a 77kb human RET sensitive haplotype into the mouse genome to generate a humanized model of Hirschsprung disease
Fine, R. D.; Low, B. E.; Rollins, J.; Laurent, J. M.; Wiles, M. V.; Zuberi, A.; Boeke, J. D.; Chakravarti, A.
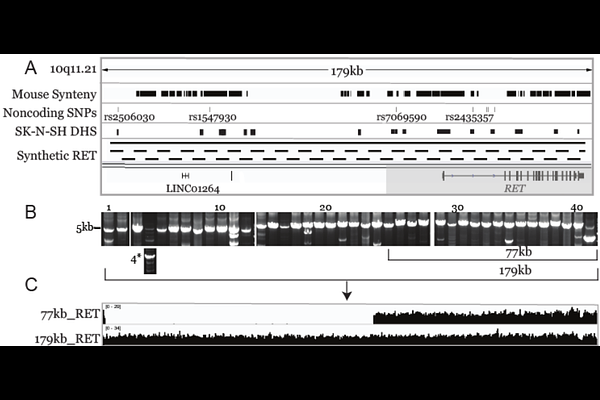
AbstractHirschsprung disease (HSCR) is a complex developmental disorder of the enteric nervous system, primarily driven by regulatory variants within enhancer elements of the RET gene. To investigate how these variants lead to aganglionosis, we developed a humanized mouse model by inserting an intact 77kb human RET genomic locus into the Rosa26 safe-harbor locus. Utilizing "big DNA" synthetic biology and Bxb1-mediated recombination, we integrated the complete human locus including all exons, introns, and upstream regulatory elements which we validated via nanopore and short-read sequencing. Functional analysis confirmed in vivo human RET expression; however, our initial HSCR-associated "sensitive" haplotype expressed at only 21% of wild-type levels. This significant reduction proved insufficient to rescue the viability when endogenous mouse Ret was deleted. We identified that this deficiency is partially driven by five risk SNPs within established enhancers. Specifically, using CRISPR/Cas9 to restore a conserved Sox10 binding site (converting a sensitive SNP to a protective one) increased RET expression by 1.9-fold and restored transcription factor binding. This study provides a robust framework for modeling human-specific regulatory disorders and demonstrates the critical impact of non-coding variation on disease pathogenesis.