Performance Evaluation of a Quantitative Metabolomics Workflow Incorporating Microchip Capillary Electrophoresis, Indexed Migration Time, and Single-Point External Calibration
Performance Evaluation of a Quantitative Metabolomics Workflow Incorporating Microchip Capillary Electrophoresis, Indexed Migration Time, and Single-Point External Calibration
Mellors, S.; Moss, C.; Redman, E. A.; Shuford, C.; Campbell, J. P.; Ramsey, J. M.; Coon, J.; Thompson, W.
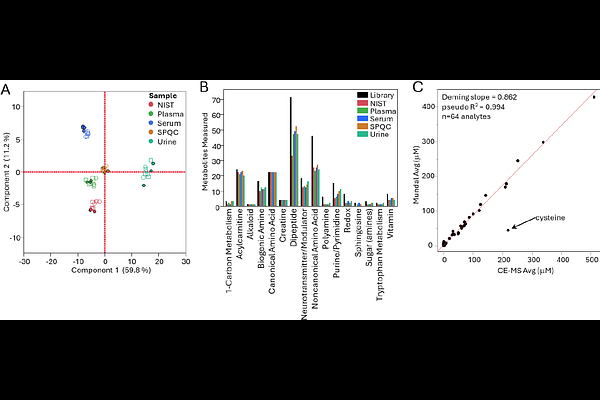
AbstractCapillary electrophoresis-mass spectrometry (CE-MS) offers unique analytical advantages for polar metabolite profiling but has remained underutilized in metabolomics relative to liquid chromatography-MS (LC-MS), in part due to challenges in managing migration time drift during data analysis. Here we introduce the use of indexed migration time (iMT) for easily managing this aspect of CE-MS data for metabolomics. Migration time indexing using a panel of stable isotope-labeled (SIL) amino acid reference standards, stored as an iRT database in Skyline, outperformed both uncorrected migration time and relative migration time (RMT) correction across three independent analytical batches spanning 90 samples from four biological matrices. The indexed migration time approach achieved sub-1% relative standard deviation (RSD) in migration index across batches, compared to up to ~15% RSD for uncorrected migration times. Additionally, we evaluate the use of single-point external calibration in Skyline for the purposes of metabolite quantification from complex matrices in order to ease the burden of translational metabolite quantification from metabolomics using high-resolution mass spectrometry (HRMS). Single-point external calibration using a biological matrix-based calibrator was benchmarked against a 13-point linear calibration curve across a panel of amino acids; above 1 uM, greater than 95% of back-calculated concentrations fell within +/-20% of multi-point calibration. Application of the complete workflow to plasma, serum, urine, and NIST Standard Reference Material (SRM)-1950 demonstrated low inter-batch variability by principal components analysis, broad metabolite coverage across 126 quantifiable analytes, and strong quantitative concordance (Deming slope = 0.862, pseudo-Rsqrd = 0.994, n = 64 analytes) with an independent comprehensive reference dataset for NIST SRM-1950. Together, these results establish a practical mCE-HRMS metabolomics workflow that bridges targeted and discovery metabolomics paradigms, and lays the groundwork for single-point external calibration as a powerful tool for translational metabolomics.