Ab initio Quantum Simulation of Strongly Correlated Materials with Quantum Embedding
Ab initio Quantum Simulation of Strongly Correlated Materials with Quantum Embedding
Changsu Cao, Jinzhao Sun, Xiao Yuan, Han-Shi Hu, Hung Q. Pham, Dingshun Lv
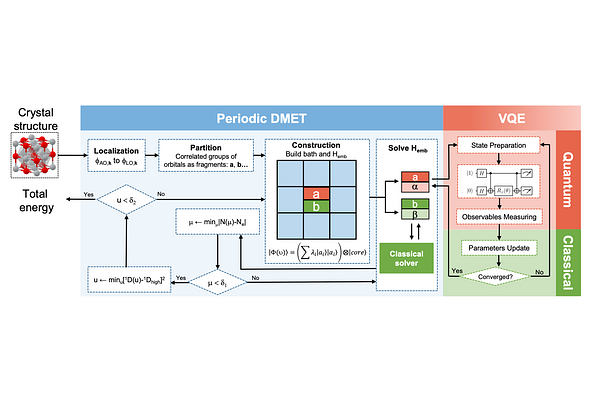
AbstractQuantum computing has shown great potential in various quantum chemical applications such as drug discovery, material design, and catalyst optimization. Although significant progress has been made in quantum simulation of simple molecules, ab initio simulation of solid-state materials on quantum computers is still in its early stage, mostly owing to the fact that the system size quickly becomes prohibitively large when approaching the thermodynamic limit. In this work, we introduce an orbital-based multi-fragment approach on top of the periodic density matrix embedding theory, resulting in a significantly smaller problem size for the current near-term quantum computer. We demonstrate the accuracy and efficiency of our method compared with the conventional methodologies and experiments on solid-state systems with complex electronic structures. These include spin polarized states of a hydrogen chain (1D-H), the equation of states of a boron nitride layer (h-BN) as well as the magnetic ordering in nickel oxide (NiO), a prototypical strongly correlated solid. Our results suggest that quantum embedding combined with a chemically intuitive fragmentation can greatly advance quantum simulation of realistic materials, thereby paving the way for solving important yet classically hard industrial problems on near-term quantum devices.