Outperforming the Majority-Rule Consensus Tree Using Fine-Grained Dissimilarity Measures
Outperforming the Majority-Rule Consensus Tree Using Fine-Grained Dissimilarity Measures
Takazawa, Y.; Takeda, A.; Hayamizu, M.; Gascuel, O.
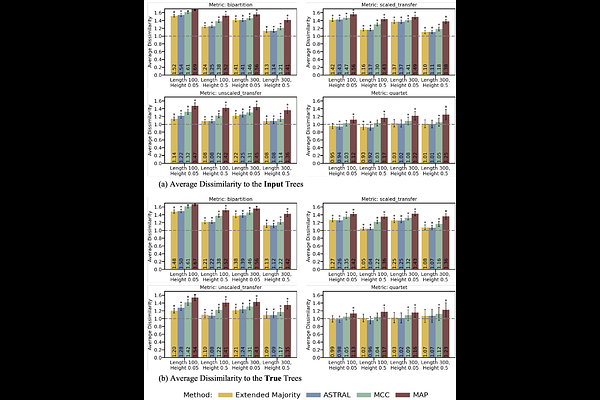
AbstractPhylogenetic analyses often require the summarization of multiple trees, e.g., in Bayesian analyses to obtain the centroid of the posterior distribution of trees, or to determine the consensus of a set of bootstrap trees. The majority-rule consensus tree is the most commonly used. It is easy to compute and minimizes the sum of Robinson-Foulds (RF) distances to the input trees. In mathematical terms, the majority-rule consensus tree is the median of the input trees with respect to the RF distance. However, due to the coarse nature of RF distance, which only considers whether two branches induce exactly the same bipartition of the taxa or not, highly unresolved trees can be produced when the phylogenetic signal is low. To overcome this limitation, we propose using median trees with respect to finer-grained dissimilarity measures between trees. These measures include a quartet distance between tree topologies, and transfer distances, which quantify the similarity between bipartitions, in contrast to the 0/1 view of RF. We describe fast heuristic consensus algorithms for transfer-based tree dissimilarities, capable of efficiently processing trees with thousands of taxa. Through evaluations on simulated datasets in both Bayesian and bootstrapping maximum-likelihood frameworks, our results show that our methods improve consensus tree resolution in scenarios with low to moderate phylogenetic signal, while providing better or comparable dissimilarities to the true phylogeny. Applying our methods to Mammal phylogeny and a large HIV dataset of over nine thousand taxa confirms the improvement with real data. These results demonstrate the usefulness of our new consensus tree methods for analyzing the large datasets that are available today. Our software, PhyloCRISP, is available from https://github.com/yukiregista/PhyloCRISP.