Erythrocytosis-inducing PHD2 mutations implicate biological role for N-terminal prolyl-hydroxylation in HIF1α oxygen-dependent degradation domain
Erythrocytosis-inducing PHD2 mutations implicate biological role for N-terminal prolyl-hydroxylation in HIF1α oxygen-dependent degradation domain
Taber, C. C.; He, W.; Gasmi-Seabrook, G. M. C.; Hubert, M.; Ferens, F. G.; Ikura, M.; Lee, J.; Ohh, M.
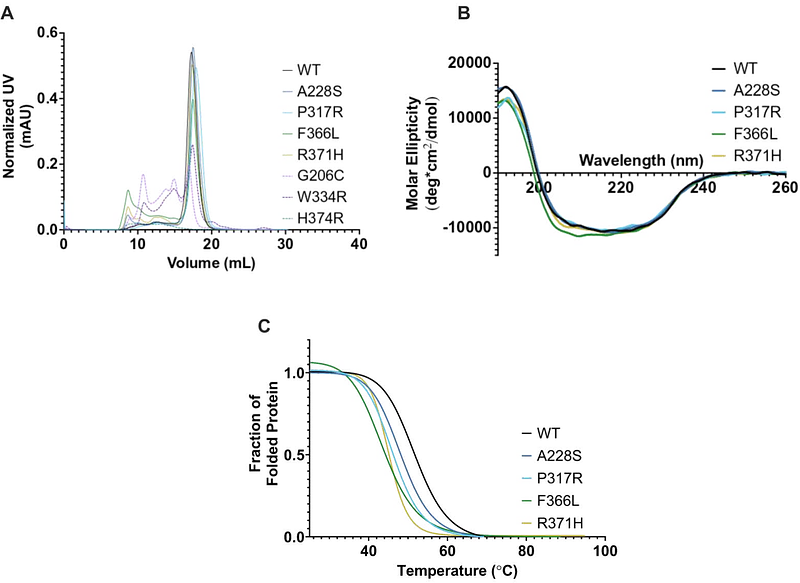
AbstractMutations in EGLN1, the gene encoding for hypoxia-inducible factor (HIF) prolyl-4-hydroxylase 2 (PHD2), cause erythrocytosis and in rare cases the development of neuroendocrine tumors. In the presence of oxygen, PHD2 hydroxylates one or both conserved prolines in the oxygen-dependent degradation domain (ODD) of HIF subunits, sufficiently marking HIF for binding and ubiquitylation via the von Hippel-Lindau (VHL) tumor suppressor protein-containing E3 ubiquitin ligase and subsequent degradation by the 26S proteasome. However, prolyl-hydroxylation in the C-terminal ODD appears to be the predominant and sufficient event in triggering the oxygen-dependent destruction of HIFa, rendering the biological significance of N-terminal ODD proline unclear. Here, we examined 7 disease-associated EGLN1 mutations scattered across the catalytic core and show definitively that all PHD2 mutants have a structural and/or catalytic activity defect as measured by time-resolved nuclear magnetic resonance. Notably, we identified one of the PHD2 mutants, P317R, to retain comparably wild-type capacity to hydroxylate the predominant proline in the C-terminal ODD but had uniquely compromised ability to hydroxylate the N-terminal ODD proline. These findings support the notion that deregulation of HIF ultimately underlies PHD2-driven erythrocytosis and challenge the currently held uncertainty that the N-terminal ODD prolyl-hydroxylation event is dispensable in normal hypoxic signaling pathway.