Abnormal ventricular wall patterning precedes and drives MYBPC3 hypertrophic cardiomyopathy
Abnormal ventricular wall patterning precedes and drives MYBPC3 hypertrophic cardiomyopathy
Salguero-Jimenez, A.; Pau-Navalon, A.; Siguero-Alvarez, M.; Relano-Ruperez, C.; Santos-Cantador, J.; Sabater-Molina, M.; Luo, X.; Lalaguna, L.; Sen-Martin, L.; Marin-Perez, D.; Galicia Martin, A.; Zhou, B.; Bernal Rodriguez, J. A.; Sanchez-Cabo, F.; Lara-Pezzi, E.; Alegre-Cebollada, J.; Gimeno-Blanes, J. R.; MacGrogan, D.; de la Pompa, J. L.
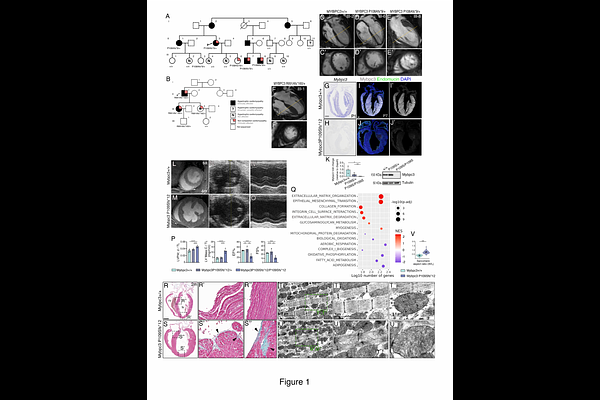
AbstractBACKGROUND: Excessive trabeculations and myocardial crypts are recurrent features across cardiomyopathies, yet their developmental origins and clinical significance remain poorly defined. To reveal the link between cardiac morphogenesis and disease, we generated humanized mouse models carrying patient-derived MYBPC3 frameshift mutations associated with overlapping hypertrophic cardiomyopathy (HCM) and left ventricular non-compaction (LVNC). METHODS: We applied CRISPR-Cas9 to introduce distinct MYBPC3 frameshift alleles into the mouse genome and performed comprehensive phenotypic and transcriptomic profiling from fetal life through adulthood. RESULTS: Adult homozygous Mybpc3 frameshift mutant mice like humans displayed hallmark HCM; however, without LVNC. Fetal and neonatal mutant hearts exhibited markedly enlarged ventricular trabeculae and crypts that progressed postnatally into the observed adult hypertrophy. Transcriptomic analysis revealed stage-specific dysregulation of oxidative metabolism, nonsense-mediated decay (NMD), and cell cycle pathways, peaking at postnatal days 1 and 7, indicating that these stages represent critical time points in disease onset. The persistent NMD signature, also observed in phenotype-negative heterozygotes, suggests a compensatory stress response. Enlarged trabeculae exhibited 2-fold increased trabecular cardiomyocyte proliferation, reversing the normal compact-trabecular proliferative gradient and leading to impaired ventricular compaction in neonates. Hey2CreERT2 lineage tracing demonstrated invasion of Hey2+ compact cardiomyocytes into the trabeculae and ectopic trabecular expression of the Prdm16 transcription factor, indicating defective ventricular wall patterning and maturation. Postnatally, Hey2+-derived cardiomyocytes became restricted to the outer/compact myocardium in mutants, while the inner/trabecular myocardium underwent accelerated hypertrophy concurrent with Prdm16 downregulation. Mice with a Mybpc3 missense variant also exhibited Hey2+ myocardial lineage expansion into trabeculae but no increased proliferation, implicating additional mechanisms beyond Hey2 regulation. Postnatal Prdm16 restoration, via transgenic expression in Mybpc3-null mice effectively attenuated hypertrophy, establishing a causal link between Mybpc3 loss, Prdm16 decline, and pathological remodeling. CONCLUSIONS: Mybpc3 governs ventricular wall maturation by regulating cardiomyocyte proliferation, patterning, and maturation, partly via Prdm16. Disruption of these developmental programs precedes and drives adult HCM, highlighting a developmental role for sarcomeric proteins, and revealing postnatal Prdm16 modulation as an antihypertrophic therapeutic strategy.