Suppression of ITPK1 and IPMK activities impairs mTORC1 signaling in pancreatic β-cells and implicates IP5 in stabilizing activated mTORC1
Suppression of ITPK1 and IPMK activities impairs mTORC1 signaling in pancreatic β-cells and implicates IP5 in stabilizing activated mTORC1
Iradukunda, C.; Salter, E. A.; Uredi, D.; Wang, X.; Wierzbicki, A.; Rameh, L. E.
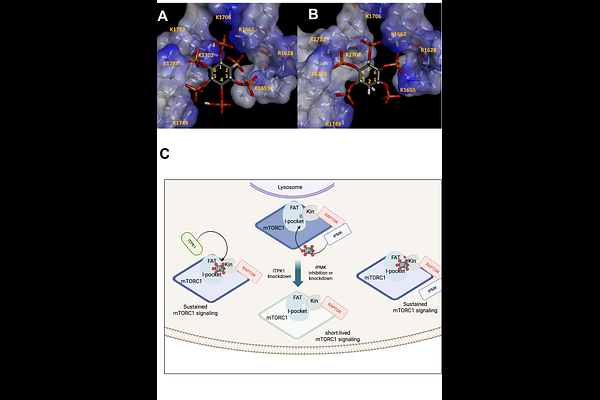
AbstractmTORC1 integrates growth factor and nutrient signals to regulate cellular metabolism, yet there are no metabolites known to directly regulate mTORC1 activity in cells. Cryo-EM studies revealed that inositol hexakisphosphate (IP6) associates with the FAT domain of mTOR, suggesting that inositol phosphates may directly modulate mTOR activity. We previously showed that higher-order inositol phosphates enhance mTORC1 kinase activity and stability in vitro. Here, we investigated whether inositol phosphate metabolism regulates mTORC1 signaling in pancreatic {beta}-cells. Suppression or acute inhibition of inositol phosphate multikinase (IPMK), as well as knockdown of inositol trisphosphate kinase 1 (ITPK1), selectively reduced cellular IP5 levels without altering IP6 and resulted in impaired basal and insulin-stimulated mTORC1 signaling, particularly under physiological glucose and low growth factor conditions. Combined inhibition of IPMK and ITPK1 nearly abolished IP5 and reduced IP6, demonstrating that these enzymes compensate to supply IP5 for IP6 synthesis. Importantly, depletion of IP5 did not impair PI3K/Akt activation but accelerated termination of the mTORC1 signal, indicating a role for IP5 in stabilizing the active mTORC1 complex. Reduction of inositol phosphate levels did not prevent insulin- or glucose-induced mTORC1 activation, revealing that IP5 primarily regulates signal persistence rather than initiation. Together, these findings identify IP5 as a metabolic regulator that prolong mTORC1 activity in {beta}-cells, providing a mechanism by which cellular metabolic state modulates sustained mTORC1 signaling.