Toward pharmacologic therapy for glioblastoma: Identifying inhibitors of very long-chain acyl-CoA synthetase 3 (ACSVL3)
Toward pharmacologic therapy for glioblastoma: Identifying inhibitors of very long-chain acyl-CoA synthetase 3 (ACSVL3)
Clay, E. M.; Shi, X.; Kolar, E. A.; Mody, M.; Locke, J. E.; Watkins, P. A.
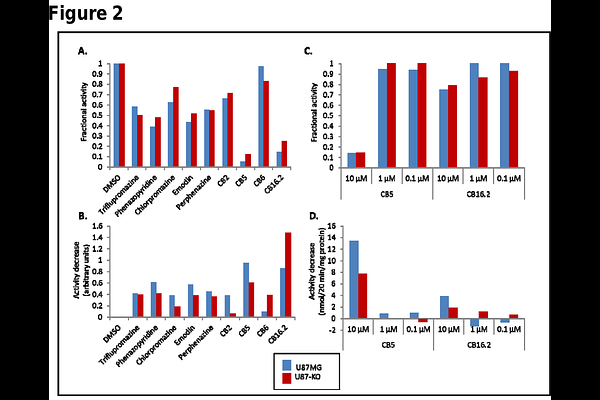
AbstractBrain tumors, in particular glioblastoma multiforme (GBM), are among the most aggressive and difficult to treat human neoplasms. Even with combined surgery, radiation and chemotherapy, the 5-year survival rate for GBM is only ~7%. Thus, new treatment approaches are needed. We previously found that the fatty acid metabolism enzyme very long-chain acyl-CoA synthetase 3 (ACSVL3) is overproduced in human glioma tissue and in glioblastoma cell lines such as U87MG cells. These cells exhibited malignant growth properties in culture and were tumorigenic in nude mice. When either knockdown or knockout strategies were used to deplete U87MG cells of ACSVL3, they adopted a more normal growth rate and produced significantly fewer, slower growing tumors in mice. An inhibitor of ACSVL3, if identified, could prove to be a valuable pharmacotherapeutic agent in GBM. Therefore, we sought to identify small molecule compounds that decrease or block the enzyme activity of ACSVL3, as measured by the formation of stearoyl-CoA from the 18-carbon saturated fatty acid stearic acid, a preferred substrate for ACSVL3. We approached this in two ways. First, we tested several compounds that were previously shown to inhibit the activity of a structurally and functionally related enzyme, ACSVL1. Several compounds tested showed inhibition of stearoyl-CoA formation in U87MG cells when added to an in vitro enzyme assay. These included drugs triflupromazine, phenazopyridine, chlorpromazine, emodin, and perphenazine which are approved for treating other conditions. Also inhibitory to stearoyl-CoA production were several compounds from a ChemBridge Corporation library designated CB2, CB5, CB6 and CB 16.2. One caveat regarding interpretation of these results is that in addition to ACSVL3, all cells including U87MG contain other acyl-CoA synthetases capable of using stearic acid as substrate. Therefore, we also measured stearoyl-CoA synthetase activity in ACSVL3-deficient U87MG cells (U87-KO). If a drug or compound is an ACSVL3 inhibitor, it should decrease total conversion of stearate to stearoyl-CoA more in U87MG than in U87-KO cells. By this criterion, most of the tested compounds showed some ACSVL3-specific inhibition. At the screening concentration of 80 M drug, CB5 and CB16.2 showed the greatest potency to inhibit ACSVL3 enzyme activity; at 10 M, CB5 still showed significant inhibition but CB16.2 did not. We conclude that these compounds are worthy of further investigation as potential therapeutic agents in GBM, but additional drugs that have greater specificity and are effective at significantly lower concentrations must also be identified. Therefore, our second strategy was to develop a high-throughput library screening assay. For this, we took advantage of the fatty acid transport capability of some ACSVL family members. ACSVL1, when heterologously expressed in COS-1 cells, promotes cellular uptake of the fluorescent fatty acid analog C1-BODIPY-C12; in contrast, overexpressed ACSVL3 does not. We used a domain-swapping strategy to replace the N-terminal 210 amino acids of ACSVL3 with the N-terminal 100 amino acids of ACSVL1, producing ACSVL1/3. Unlike ACSVL3, ACSVL1/3 robustly promoted C1-BODIPY-C12 uptake while retaining the catalytically active C-terminus of ACSVL3. Most of the drugs and compounds that decreased stearoyl-CoA synthetase inhibition also inhibited C1-BODIPY-C12 uptake in a concentration-dependent manner. Catalytically defective ACSVL1/3 mutants lost their ability to promote C1-BODIPY-C12 uptake. Thus, we conclude that chimeric ACSVL1/3 gained the fatty acid transport function of ACSVL1 while retaining the catalytic properties of ACSVL3. A pilot screening study of >1280 drugs from an approved drug library and >880 compounds from a library of drugs predicted to cross the blood-brain barrier detected more than 50 molecules that lowered C1-BODIPY-C12 by more than 3 standard deviations. Although secondary screening will likely exclude many or all of these, our findings support the notion that we have developed a viable method for detecting potential ACSVL3 inhibitors. Further characterization may reveal candidate pharmacologic agents for treatment of GBM and other cancers.