Long-Read Transcriptome Sequencing and Functional Validation Reveals Novel and Oncogenic Gene Fusions in Fusion Panel-Negative Gliomas
Long-Read Transcriptome Sequencing and Functional Validation Reveals Novel and Oncogenic Gene Fusions in Fusion Panel-Negative Gliomas
Rybacki, K.; Cha, E. N. Y.; Deutsch, H. M.; Gaudet, E.; Ahsan, M. U.; Xu, F.; Chan, J.; Li, M.; Song, Y.; Wang, K.
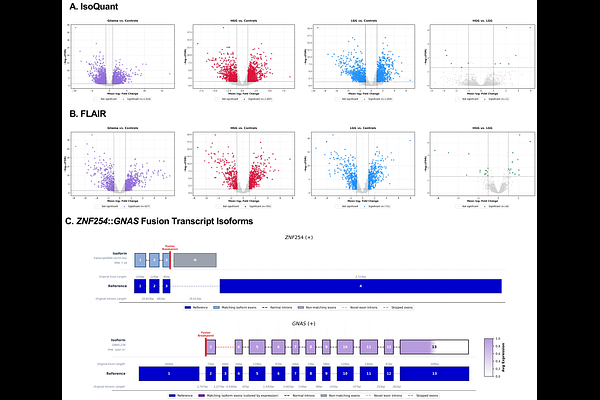
AbstractGliomas comprise a heterogeneous group of central nervous system tumors in which gene fusions (GFs) are significant oncogenic drivers and emerging diagnostic and therapeutic biomarkers. In cancer diagnosis, GF detection largely relies on targeted short-read sequencing fusion panels, such as the Children's Hospital of Philadelphia (CHOP) Fusion Panel (FUSIP). While these panels are effective for detecting recurrent, well-characterized GFs, they are limited to predefined gene sets and cannot identify full-length transcripts. Here, we analyzed 49 high- and low-grade gliomas previously classified as fusion-negative by FUSIP using an untargeted whole-transcriptome RNA sequencing approach with Oxford Nanopore Technologies (ONT) long-read sequencing. This enabled transcriptome-wide fusion discovery of additional known and potentially novel oncogenic GFs beyond panel constraints. Long-read sequencing further allowed direct resolution of full-length fusion transcripts and their associated isoform structures. By integrating GF detection with isoform-level transcript analysis, we identified fusion-associated transcript isoforms with alternative splicing patterns that aligned near reported GF breakpoints, including ZNF254::GNAS and PTPRK::NOX3, which have not been reported in literature or existing fusion databases. To assess functional relevance, candidate GFs were evaluated using the Drosophila melanogaster model, with ventral nerve cord (VNC) morphology serving as a quantitative in vivo readout of fusion-induced disruption of glial regulation. VNC enlargement or elongation reflects abnormal glial growth or defects in brain tissue organization. Of the 15 candidate GFs subjected to experimental functional testing, 8 induced significant VNC abnormalities relative to wild-type controls, indicating fusion-specific disruption and oncogenic potential. Notably, CLDND1::WRN and DUSP22::APOE produced the most pronounced VNC phenotypes. Together, these findings demonstrate that untargeted transcriptome-wide GF discovery, coupled with long-read isoform-level analysis and in vivo functional validation, enables the identification and prioritization of potentially novel and clinically relevant GFs that are missed by standard targeted short-read fusion panels in glioma.