Sampling and ranking of protein conformations using machine learning techniques do not improve quality of rigid protein-protein docking
Sampling and ranking of protein conformations using machine learning techniques do not improve quality of rigid protein-protein docking
Kyrylenko, R.; Koleiev, I.; Savchenko, I.; Voitsitskyi, T.; Stratiichuk, R.; Husak, V.; Yesylevskyy, S.; Starosyla, S.; Nafiiev, A.
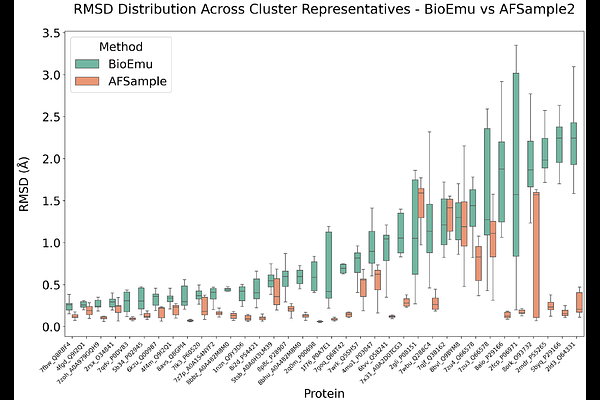
AbstractRigid docking remains the most popular method of predicting the protein-protein interactions in the case when experimental 3D structures of the complexes are not available. The docking often rely on known unbound (Apo) protein structures, which may differ significantly from their bound (Holo) forms. Modern machine learning (ML) based conformational sampling techniques allow generating ensembles of functionally relevant protein structures, which may be closer to their Holo forms and thus could improve the outcomes of the classical rigid protein-protein docking. Here we sampled conformations of the protein subunits in 30 complexes from the novel PLINDER dataset with two state-of-the-art ML-based techniques and evaluated their docking performance using several physics-based, data-based and ML-based scoring functions. We showed that such conformational sampling rarely produces structures that are closer to the Holo conformations than the corresponding Apo ones. Moreover, even when such conformations are generated, none of the tested scoring functions were able to prioritize and rank them correctly. Our work highlights critical limitations in the current ML-enhanced rigid protein-protein docking workflows and emphasizes the need for new approaches that can better utilize the potential of modern techniques for conformational generation and scoring.